

Definition
The retinoblastoma (RB) is a highly malignant tumour of the retina, and its prognosis depends on an early diagnosis and treatment.
Cause
This tumour is due to a mutation in RB1 gene, and its development derives from the existence of a mutation in the two copies of the gene. The first mutation may involve germ cells or it may be confined to the tumour itself. This difference determines two distinct forms of the disease: hereditary RB and non-hereditary RB. These two forms of tumour present very different clinical and epidemiological characteristics.
GENETIC CLINIC
Hereditary retinoblastoma
Only 10% of the patients with retinoblastoma have a history of this disease within the family. But a new mutation in germ cells originates nearly more 30% of cases. Thus, about 40% of the patients have a hereditary tumour.
These patients present a mutation in RB1 gene in all the cells of the organism: a germinal mutation. It may be inherited from a parent or, more frequently, it may occur in a cell in the embryonic stage of the individual. The mutation in the second copy of the RB1 gene occurs in a multipotential retinal cell that originates the tumour.
The RB1 gene has a recessive behaviour and so the existence of a tumorous phenotype implies that both alleles are affected. However, at an individual level, the mutation behaves in an autosomal dominant way, with a penetration of 90%.
The patients with this hereditary form of disease present a high risk, from 80% to 90%, of developing a bilateral or multifocal ocular tumour during infancy; furthermore, they have a predisposition to develop non-ocular tumours throughout their life. About 8% of them develop a second primary malignant intracranial tumour, generally in the pineal gland – trilateral retinoblastoma. About 30% of these patients develop another non-ocular tumour before they are 40 years old. The most frequent being the osteosarcomas, the fibrosarcomas and the melanomas.
Non-hereditary retinoblastoma
The remaining 60% of the patients with retinoblastoma have a non-hereditary or somatic form of the disease. In these patients the two mutations in RB1 gene occurred in only one retinal cell, originating a unilateral and unifocal form of the tumour. These patients have no predisposition to develop non-ocular tumours.
Epidemiology
Retinoblastoma is the most frequent ocular tumour in children. It occurs approximately in 1 out of every 20,000 children and it represents about 12% of all infancy tumours.
The average age at diagnosis is 13 months in bilateral forms of the disease and 24 months in unilateral forms. In patients screened because of familial precedents, the diagnosis is naturally made at an earlier age, in many cases shortly after birth.
About 2/3 of the tumours develop before the age of 2 years and 95% develop before the age of 5 years. Over the age of 7 years, these tumours are very rare.
Clinical aspects
The diagnosis of retinoblastoma is not equally easy in families affected by it and in families without a history of this disease. Among the children of parents with retinoblastoma, the diagnosis is usually more precocious, in a stage when the tumour is smaller and offers a better opportunity of treatment to preserve the eye and sight.
> Signs and symptoms
In case of no history of retinoblastoma within the family, the tumour is diagnosed by accident or after detecting an alteration in the eye.
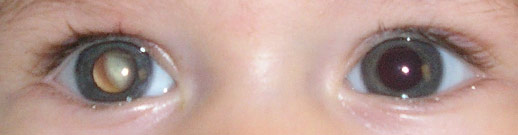
The most common sign of existence of the tumour is leucocoria (white pupil), in 60% of the cases. The second most frequent alteration is the development of a strabismus. Other more rare symptoms include: intraocular inflammation, neovascularization of the iris, hyphema (blood in the anterior chamber) orbital cellulitis and glaucoma.
In developed countries, the disease seldom presents evidence of extraocular extension, metastatic development, or signs of involvement of the central nervous system.
> Growth of the tumour
If not treated, the tumour will grow, filling up the eye socket and eventually spreading itself out of the eye.
The retinoblastoma presents four forms of growth. The endophytic pattern is characterized by a growth towards the vitreous cavity, making the diagnosis easier because of direct visibility. In the exophytic pattern, there is a growth towards the choroid, raising the retina as in a serous detachment. Generally, most of the tumours present characteristics common to these two patterns of growth – mixed pattern. The fourth pattern of growth is a diffuse form, quite rare (it represents 1% to 2% of the cases), in which the tumour infiltrates the retina producing a thickening in plaque: clinical signs are very subtle and many times the diagnosis is delayed.
Retinoblastoma cells are not very cohesive and they frequently spread to the vitreous humour (vitreous sowing) or to the sub-retinal space, depending on the direction of the growth of the tumour. They may also disseminate to a site distant from the primary tumour and the retina itself (mimicking a polycentric tumour), to the crystalline capsule or even to the anterior chamber (with a possible complication of glaucoma or uveitis).
> Differential diagnosis
In typical cases, the diagnosis is easy. But there is a significant number of cases in which the diagnosis of retinoblastoma is not confirmed. The diseases that lead more times to a wrong diagnosis of retinoblastoma are: persistence of foetal vascularization (PFV), Coats’ disease and toxocara inflammation (granuloma).
> Ophthalmological evaluation
In case of suspicion of existence of retinoblastoma, the ophthalmologist should evaluate carefully the visual capacity of the affected eye or eyes. This is a determinative issue for future therapeutic decisions.
The ophthalmological examination should be as detailed as possible, according to the age and the cooperation of the child, and it should include the search for signs associated with the existence of the tumour.
The observation of the tumour should be made with dilation of the pupil, in order to allow a careful examination of all the eye fundus including the periphery of the retina. Whenever possible, a colour photographic record of the tumour should be obtained with a contact camera and a wide-angle lens. This record is fundamental for debating therapeutic strategies and for monitoring the response to therapies.
> Auxiliary exams for diagnosis
Ultrasonography is an extremely useful instrument in the evaluation of retinoblastoma.
In mode A, it is possible to record high intensity echoes inside the tumour, corresponding to calcification areas. The mode B of the echography demonstrates the presence of intraocular tissue, through highly reflective echoes dispersed inside the tumour and through a sign toned down behind the tumour. It also allows diagnosing the existence and the extent of associated retinal detachment.
Computerized tomography and magnetic resonance imaging confirm the diagnosis and enable an evaluation of the extraocular extension of the tumour. The tomography is particularly sensitive to scrutinize intratumorous calcifications characteristic of the retinoblastoma, to evaluate the extension of the tumour outside the eyeball, and to exam the orbit and contiguous bone structures.
Magnetic resonance imaging is particularly important to evaluate the extension of the tumour along the optic nerve and in the central nervous system.
In most of the cases, the exhaustive search for metastases is reserved for patients presenting characteristics of very advanced disease. It is always obligatory to request an evaluation by a paediatric oncologist.
Treatment
The treatment should be individualized for each patient. It is determined by several factors including the size and position of the tumour, the visual capacity of the affected eye, the involvement of the optic nerve, the expansion to the orbit and the existence of metastases.
Briefly, in case of small tumours (size inferior to 3 millimetres) only local treatments are applied, with laser or with cryotherapy. For larger tumours and especially for multiple or bilateral tumours, it is preferable to use chemotherapy to reduce the size of the tumour (chemo-reduction) and to apply local treatments only afterwards, with laser therapy, cryotherapy or radiotherapy. In very extensive tumours, generally involving more than 50% of the eyeball, or in tumours with opacity of transparent structures or neovascularization of the iris, the solution is the enucleation of the eye.
The priority of the treatment of retinoblastoma is to save the life of the child, but the great evolution of the therapies in the last decades also enables, in most of the cases, to preserve the eye affected by the tumour and to ensure the best possible visual function.
Medical surveillance after treatment
The hereditary retinoblastoma implies a high risk of onset of other retina tumours during childhood. Thus, a close medical surveillance is necessary throughout this period of life. In addition, the familial form is associated with the onset of other malignant tumours during lifetime. The frequency of checking examinations should be individualized, involving necessarily multidisciplinary teams and recurring all lifelong.
Prognosis
The prognosis concerning survival is now excellent, and ocular and visual morbidity is also much reduced. In developed countries, mortality rate has decreased to values below 10%.
The long-term prognosis is worse for hereditary forms of the disease, because of the predisposition to develop extraocular tumours, and so these cases require a more careful medical surveillance.
The visual function of the patients improved significantly with the development of treatments that preserve sight and avoid enucleation.
Despite the great improvement of the prognosis, metastatic retinoblastoma is still difficult to treat and its precocious detection is an extremely important factor.
In case of pineal gland involvement, the prognosis is very bad, with a mortality rate of nearly 100% at the age of 5 years.